
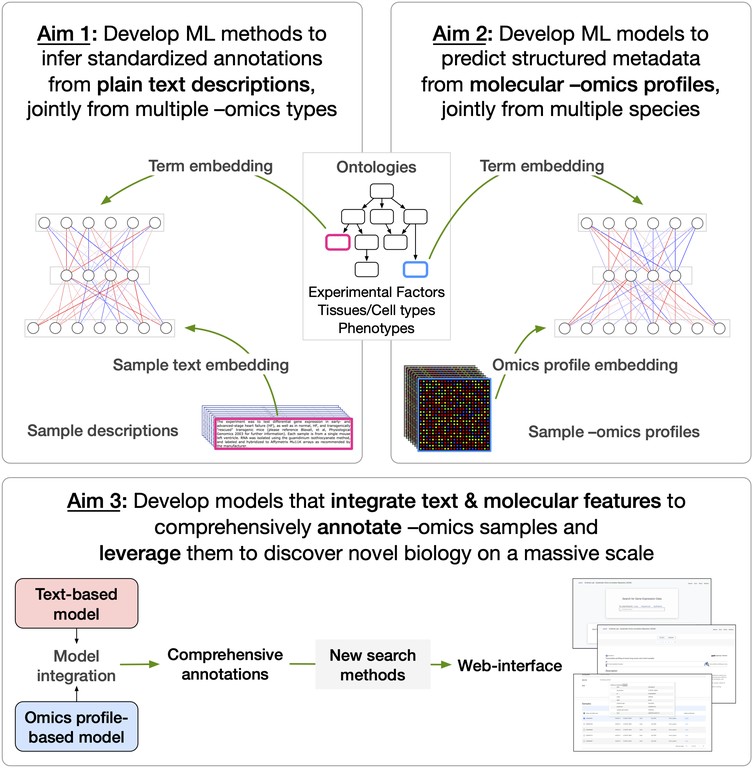
In our project, we are developing new machine learning (ML) methods to assign comprehensive, standardized annotations to nearly 2 million publicly-available omics samples to promote their effective reuse by the biomedical community. We are developing:
We are a diverse, collaborative academic research group consisting of one faculty member, one postdoc, two graduate students, one undergraduate researcher, and one programmer. We are part of two departments (Computational Math, Science and Engineering; Biochemistry and Molecular Biology) and one service unit (Data Management and Analytics) at Michigan State University. The faculty member and postdoc take the lead on defining the questions, setting long- and short-term goals, overseeing progress, and providing timely mentoring and feedback. The graduate students work with the undergrad and the postdoc to develop the computational methods, which includes conceptualization, implementation using Python/R, data download/organization/processing, running algorithms on the high-performance cluster, and summarizing and interpreting results. Some of the graduate students and the postdoc also take the lead in reviewing, organizing, documenting, and disseminating the final software and data that enable other researchers to reproduce and extend our work. The programmer takes the lead in developing an interactive web-server that biologists can use to run our method and query, visualize, and export our results in a variety of convenient formats.
Started in 2018, our project’s overarching goal is to assign comprehensive, standardized annotations to millions of publicly-available omics samples to enhance the ability of the whole biomedical community to discover, reuse, and interpret these published data. We have set out to achieve this goal by:
We have developed the first versions of the two methods and shared them with the community as an open-source repository github.com/krishnanlab/txt2onto. The repo contains the pre-trained NLP-ML models and a Python utility called Txt2Onto for text-based sample annotation of new samples based on their descriptions and to train new custom text-based NLP-ML models based on user-defined training data.
A compelling aspect about our sharing/reuse practices is our effort to use our pre-trained models to add tissue annotations to hundreds of thousands of existing omics samples from human and mouse. The annotations are to terms in a controlled vocabulary of tissue and cell type terms in the UBERON and Cell Ontology. We are now working with a software developer to build a queryable web-interface Meta2Onto where a researcher can go to search for their sample attribute of interest (tissue, disease, etc.) and get back a comprehensive list of public omics datasets and samples with that attribute.
A practice we are implementing — and recommend to all researchers adopt — is the creation of fully reproducible open case studies that document (with data, code, description, and screencast) the application of ML-based methods to specific biomedical problems/datasets. These case studies — along with documentation of known and potential limitations of various aspects of data, models, and prediction — are critical for showcasing the application and improving the transparency and trust of data-driven ML research in biomedicine
As mentioned above, we shared our methods with the community as an open-source repository github.com/krishnanlab/txt2onto centered on a Python tool for text-based sample annotation called Txt2Onto. In addition to providing open, well-documented, modular code and pre-trained ML models, we invested effort in building Txt2Onto specifically to enable any researcher to not only replicate our approach but also to easily extend it.
Researchers can use Txt2Onto to do two things:
We have provided demo scripts and detailed instructions that outline how to use and extend the tool. This is an approach that we recommend to other researchers: invest effort not just in depositing code but in demonstrating clearly how the code can be used for various applications.
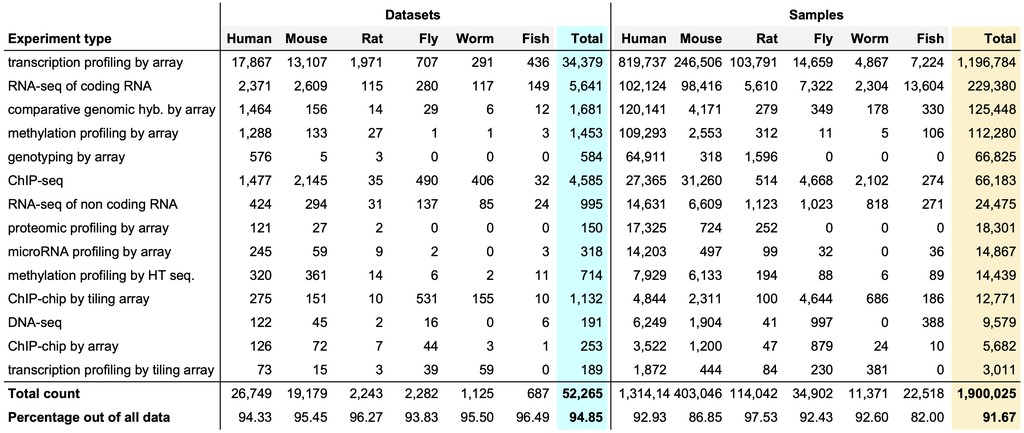
Currently, there are nearly 2 million omics samples and >52,000 omics datasets that are publicly available in repositories like the ArrayExpress. The majority of these data are from six major organisms used in biological research: human, mouse, rat, fly, worm, and fish. Data generated using the top 15 most common types of high-throughput experiments account for nearly 92–95% of all omics data.
These ~2 million samples capture large-scale cellular responses of diverse tissues and cell types in human and model organisms under thousands of different conditions, making these published omics data invaluable for researchers to reuse to:
However, given how inconsistent and incomplete sample annotations currently are, biologists who wish to use this valuable resource have to spend days, weeks, or even months finding and curating relevant published data, which takes away from devoting their resources to gleaning novel biology. We are working on removing this barrier.
