AA's team
We demonstrate how publicly shared single-cell RNA-seq datasets can be reused to predict gene perturbation outcomes without new experiments. Focusing on human T cells, we addressed key reuse challenges: incomplete metadata, unclear provenance, and heterogeneous repository formats. We defined environments by combining tissue, donor, and perturbation metadata, then curated and quality-controlled datasets while preserving biological variation across environments. Leveraging these natural differences, we applied Anchor Regression to learn causal gene regulatory networks, which enable in-silico knockout predictions with quantified uncertainty and optimization of multi-gene combinations for cell engineering. We release all processed data, analysis code, and an interactive web app. Our framework shows that systematic integration of existing data can yield mechanistically interpretable models that provide cost-efficient hypothesis generation for therapeutic design.
Predicting gene-perturbation outcomes from existing data expands biological insight without new experiments. Exploring all combinatorial perturbations in the lab is infeasible; computational screening helps by (1) mapping causal interactions among genes to reveal mechanistic biology and (2) identifying interventions that shift transcriptional states toward desired outcomes, guiding therapeutic strategies.
When a relationship between two genes stays consistent across datasets and conditions—what we define as environments—it is likely causal rather than correlational. Each environment (tissue, donor, microenvironment) introduces natural variation that acts as a shift in gene expression. Capturing these shifts allows us to distinguish genuine biological mechanisms from context-specific noise. Testing across many environments strengthens the inference that invariant gene-gene links represent causal regulatory edges.
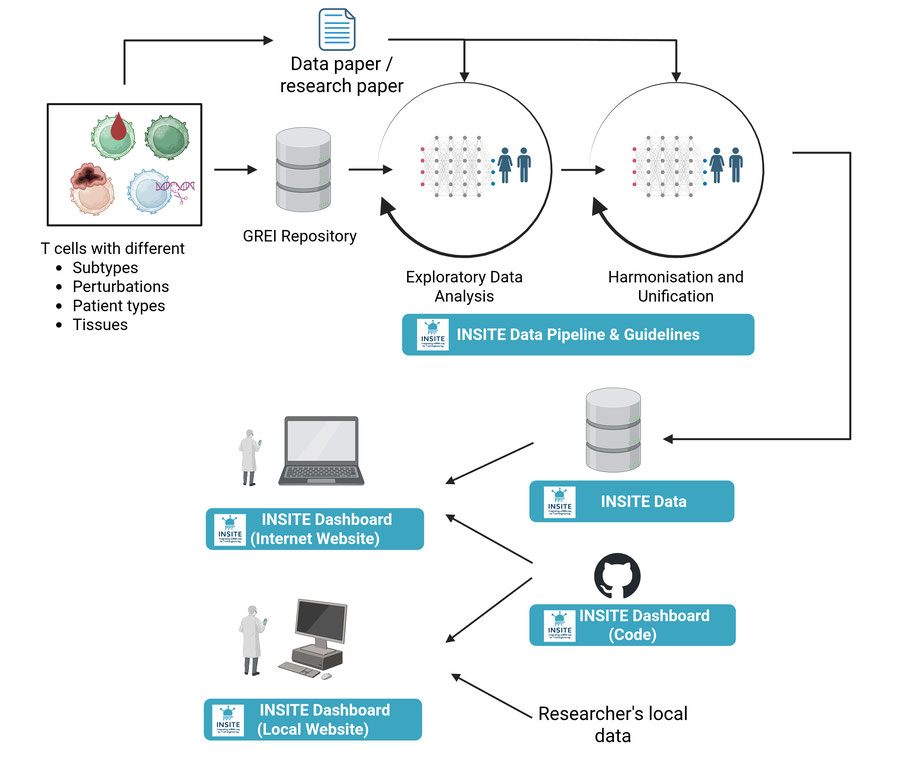
We reused public single-cell RNA-seq datasets of human CD4 and CD8 T cells from GREI repositories (Zenodo and Figshare). Because cell types and environments were inconsistently labeled, we standardized environment definitions by combining perturbation, donor, and tissue metadata into explicit labels (e.g., lung tumor, healthy, or CRISPR knockout [KO]). For each record we generated one h5ad file per distinct environment.
We curated metadata, performed QC, removed technical artifacts, and retained biological differences across environments, treating them as natural shift interventions to learn stable gene-gene relationships. Deliverables include a data model, Python library implementing the pipeline, and static website showcasing three core functions detailed below.
Explore. Provides harmonized embeddings (e.g., UMAP) and differential-expression analysis between any two environments for interactive comparison.
Predict. Unlike black-box deep-learning methods for KO prediction, we construct an interpretable gene regulatory network (GRN) represented as a structural causal model and learned through Anchor Regression. This approach estimates per-gene relationships while penalizing residual variation linked to environmental factors. Leveraging causal-inference principles (do-calculus), we conduct in-silico perturbations to predict mean and distributional changes across genes, enabling statistical testing (p-values and FDR) for KO versus baseline conditions.
Optimize. Given a target transcriptional profile and a KO budget, the tool searches for multi-gene combinations predicted to move the cell toward the goal, illustrating applications to rational cell-engineering design.
We identify environment-invariant regulatory edges that enable in silico perturbation prediction with quantified uncertainty and support gene-wise differential-expression testing for KO versus baseline across environments.
Existing single-cell data, when systematically organized across well-defined environments, can be reused to infer GRNs that generalize to unseen perturbations—providing a cost-efficient and mechanistically interpretable framework for hypothesis generation.
All single-cell datasets used were sourced from Zenodo and Figshare. Every record was processed independently to compute normalized, residualized expression matrices; environments were defined from metadata and records partitioned accordingly. We also incorporated PerturBase T-cell datasets (13 datasets, ~200k cells). Our processed dataset is publically released here: https://zenodo.org/records/17283912.
Can we learn T-cell GRNs from pre-existing data and predict KO effects with uncertainty?
Approach: Treat environments as shift interventions and apply Anchor Regression to identify stable gene-gene relationships. Resulting networks yield per-environment KO predictions with mean shifts and confidence intervals reflecting immune circuitry.
Can we identify effective combinatorial KO for cell engineering via in-silico causal models?
Approach: Using the learned causal network and set function optimization, we find feasible multi-gene KOs that move expression profiles toward user-specified targets, demonstrating a pathway from causal inference to translational design.
Knockouts are a foundational way scientists learn how cells work—by removing a gene and observing what breaks, what compensates, and what rewires. In T cells, the stakes are especially high. We care not only about single-gene effects but also about combinations that can redirect fate, amplify desirable programs, or release cells from exhaustion. Exhaustion is a dysfunctional state that blunts T-cell responses during chronic infection and cancer; lifting it is central to modern immunotherapy. Our work shows that large, heterogeneous single-cell datasets—already in the public domain—can be reused as natural experiments to infer environment-robust gene–gene relationships and to predict the consequences of knockouts before going back to the bench.
Scientifically, the project contributes a practical template for data reuse at scale in immunology: extract cell types and environments buried across studies, retain biological context rather than harmonizing it away, and learn environment-invariant regulatory controls. From those learned regulatory rules, we compute both average and distributional KO outcomes, which yields not just point predictions but also uncertainty and differential expression (DE) evidence. This moves beyond static atlases toward a predictive atlas—one that can prioritize perturbations and combinations under a KO budget to approximate desired transcriptional states.
For human health, the immediate impact is on treatment development. In oncology, our in-silico KO screening can help nominate targets and multi-target combinations to push tumor-infiltrating T cells away from exhaustion and toward durable effector or memory-like programs—conceptually synergistic with checkpoint blockade and adoptive cell therapies (such as engineered T cells). In infectious disease, the same approach can propose perturbations that enhance antiviral or antibacterial responses without tipping cells into exhaustion, informing adjuvant and regimen design. In autoimmunity, it provides a way to explore suppressive combinations that might selectively dampen pathogenic T-cell programs while preserving host defense. For vaccines, KO-guided hypotheses can highlight regulatory axes that bias toward effective memory formation.
Because our framework quantifies uncertainty and reports DE genes per environment, it supports comparative evaluation across tissues, donors, and disease contexts—an important step toward translational relevance. While our primary influence is on treatment strategy (target nomination and combination design), the same network-level readouts can surface candidate biomarkers of response or resistance (impact on diagnosis) and suggest directions for rational adjuvanting (contributing to prevention).
The broader impact is a shift in practice: using public single-cell resources not only to describe cell states, but to forecast the effect of perturbing them—accelerating hypothesis generation and focusing scarce experimental effort where it most matters.
Completion within award period
Yes. We delivered per-environment CD4/CD8 files, GRN learning with Anchor Regression, in-silico KO (mean and distribution), DE testing, and public releases of code, data artifacts, and docs.
Scope changes
Constraints and mitigations
a) Methods to validate quality & completeness
b) Issues encountered
Reusing heterogeneous research data presented several challenges that we systematically addressed. Many datasets lacked consistent metadata, documentation, or standardized formats, making it difficult to integrate them into a unified framework. To overcome this, we developed semi-automated curation tools combined with manual triage following human-in-the-loop guidelines to assess data completeness, provenance, and compatibility. Another major challenge was reconciling differing gene identifiers, annotation systems, and naming conventions across repositories; this was mitigated by implementing mapping scripts and cross-checking against authoritative reference databases
On the analytical side, variability in preprocessing pipelines, sequencing depth, and quality metrics required harmonization before comparative analysis. We employed batch correction and normalization procedures, coupled with robust quality control metrics from established frameworks such as scanpy, to ensure consistency.
