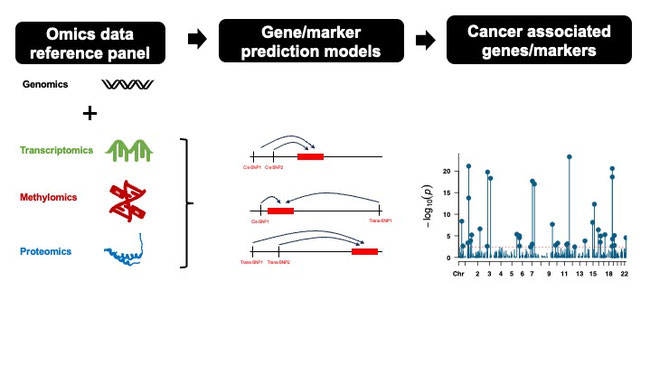
The incidence and mortality rates of many cancers vary significantly across different racial and ethnic groups, yet identifying causal biomarkers remains challenging. This project aims to uncover putative causal genetic, epigenetic, and protein biomarkers for multiple cancer types by integrating the largest available multi-omics and GWAS datasets across diverse populations. By leveraging genetic instruments to estimate genetically regulated components of gene expression, DNA methylation, and protein levels, we will mitigate common limitations of traditional observational studies. Our approach combines state-of-the-art methods including multi-population meta-analysis, cross-cancer analysis, Mendelian Randomization, and machine learning. This comprehensive strategy will help to generate crucial insights into cancer etiology across populations, enable precision risk prediction, inform targeted prevention strategies, and significantly improve health disparities.
Cancer remains a leading cause of mortality worldwide, with incidence and mortality rates exhibiting significant variability across different racial and ethnic populations. Despite extensive research, identifying causally relevant biomarkers remains challenging, particularly for non-European populations. Traditional observational studies are often limited by selection bias and confounding.
To address these gaps, we propose a comprehensive multi-omics study to identify putative causal biomarkers for cancer risk across diverse populations. We will integrate multiple data resources, many of which are deposited in GREI repositories and contributed by us:
Our innovative approach employs TWAS, MWAS, and PWAS to predict gene expression, DNA methylation, and protein levels using genetic instruments and assess associations of predicted marker levels with cancer outcomes. This strategy mitigates selection bias and unmeasured confounding. To enhance robustness and generalizability, we will implement: 1) Multi-ethnic meta-analysis for population-specific and pan-population biomarkers; 2) Cross-cancer analysis for common biomarkers; and 3) Mendelian Randomization for causal inference.
Timeline:
Our project aims to generate a comprehensive catalog of putative causal genetic, epigenetic, and protein biomarkers for multiple cancer types across diverse populations. We anticipate that this research will yield significant insights into shared biological pathways across several cancer types and enable the development of improved risk prediction models that incorporate multi-omics data. Furthermore, our findings may unveil potential novel therapeutic targets for future investigation.
To maximize the impact of our research, we will disseminate our findings through multiple channels. Primary results will be published in peer-reviewed journals, with a preference for open-access options to ensure widespread accessibility. Our project is designed to adhere to FAIR principles, ensuring that our data and findings are Findable, Accessible, Interoperable, and Reusable. All relevant data, including associations summary statistics and analysis scripts etc, will be submitted to osf.io, leveraging its robust infrastructure for long-term data preservation and sharing. To promote Reusability, we will provide detailed data dictionaries, quality metrics, and provenance information for all datasets. Given our focus on racial and ethnic diverse populations to ensure benefits for all, we are committed to ensuring Collective Benefit, Authority to Control, Responsibility, and Ethics (CARE principle) in our research.
To address replicability and reproducibility, we will publish detailed methods and analysis protocols alongside our results. All codes used in our analyses will be made available on GitHub with appropriate version control, including specific commit hashes for exact reproducibility. We will utilize containerization technologies such as Docker to ensure computational environment reproducibility, providing Dockerfiles and container images. Comprehensive documentation of all data processing steps, including software versions and parameter choices, will be provided in a structured format (e.g., Jupyter notebooks) to enable other researchers to replicate our findings.
By implementing these rigorous standards for data sharing, ethical considerations, and reproducibility, we aim to maximize the long-term impact and utility of our research findings. This approach will ensure that our results are not only widely accessible but also reusable and reproducible by the broader scientific community, thereby advancing our collective understanding of cancer biology across diverse populations and potentially informing future personalized prevention and treatment strategies.
Our research project stands to make substantial contributions to cancer genomics, epidemiology, and precision medicine, while potentially revolutionizing cancer diagnosis, treatment, and prevention strategies across diverse populations.
In the realm of scientific advancement, our study will pioneer the integration of multi-omics data (transcriptomics, epigenomics, and proteomics) with genetic information across multiple cancer types and diverse racial & ethnic groups. This comprehensive approach will help elucidate the complex interplay between genetic and environmental factors in carcinogenesis, significantly enhancing our understanding of cancer biology. The novel application of TWAS/ MWAS/PWAS in a multi-population, multi-cancer context will not only advance statistical genetics methodologies but also address the critical issue of population bias in genomic studies.
The potential clinical impact of our research is profound. The identification of causal biomarkers across diverse populations could enable the development of population-specific risk prediction models with substantially improved accuracy. These enhanced models could dramatically improve cancer screening protocols, enabling earlier detection and intervention, particularly in high-risk individuals across different racial/ethnic groups. Our findings may also uncover novel therapeutic targets, potentially catalyzing the development of innovative, population-specific cancer treatments.
In the arena of prevention, our research could transform cancer risk stratification. The identification of causal, population-specific biomarkers will allow for more targeted prevention strategies, including tailored lifestyle interventions or chemoprevention approaches based on molecular mechanisms. Our cross-cancer analysis will reveal shared causal pathways, potentially identifying common prevention strategies effective across multiple cancer types. This personalized approach to cancer prevention could significantly reduce cancer incidence rates across diverse populations.
In summary, we create a foundation for systematic evaluation of cancer risk across diverse groups. This approach will reduce healthcare disparities through improved biological understanding and targeted interventions, enable development of targeted therapies, and inform population-specific prevention strategies, ultimately advancing health equity in cancer care. Furthermore, our reproducible analytical pipelines will accelerate the conduction of similar studies for other diseases, multiplying the impact of our methodological innovations.
Our research team comprises two highly accomplished investigators who bring complementary expertise to this project: Dr. Chong Wu, an Assistant Professor of Biostatistics at The University of Texas MD Anderson Cancer Center, and Dr. Lang Wu, an Associate Professor and Director of Pacific Center for Genome Research at University of Hawaii.
Dr. Chong Wu is a biostatistician with extensive experience in statistical genetics and machine learning. His innovative work in integrating multi-omics data has led to publications in prestigious journals such as Nature Communications, Annals of Statistics, and Biometrics. His expertise in developing novel statistical methods is crucial for our project's aim to identify causal biomarkers across diverse populations and cancer types.
Dr. Lang Wu is an experienced genetic and cancer epidemiologist studying cancer health disparity. His groundbreaking omics integration work published in high impact journals such as Nature Genetics demonstrates his proficiency in applying advanced statistical methods to large-scale genomic and non-genetic data.
Our team's collaboration spans over five years, resulting in over 30 co-authored publications. This long-standing partnership has fostered a synergistic relationship, seamlessly integrating our expertise in biostatistics, genetic epidemiology, and cancer research. Our complementary skills and established collaboration provide a strong foundation for executing this innovative and impactful research project.
The success of our research project depends on:
Data Quality and Integration: High-quality, harmonized multi-omics data across diverse populations will be ensured through stringent quality control and standardized integration protocols.
Statistical Methodology: Robust statistical methods are central to our project. Our extensive expertise in statistical analysis of multi-omics data will ensure the success of our research.
Collaboration and Communication: Maintaining effective collaboration between team members is vital. We have established regular team meetings and progress reviews to facilitate seamless coordination.
Reproducibility: Ensuring reproducible findings is vital for long-term impact. We will use version control for all analyses, containerize computational environments, and provide comprehensive documentation of our methods.
By carefully addressing these considerations, we aim to maximize the reliability, impact, and translational potential of our research findings.
