CRISPR gene editing holds great promise for therapeutic use but is challenged by off-target effects, which can cause unintended mutations, including those with oncogenic potential. This project aims to enhance the safety of CRISPR-based therapies by analyzing two previously unexplored population variants datasets (All of Us (AoU) and gnomAD) which together encompass over 1.5 billion novel variants. By integrating these datasets with cancer driver mutations from Mendeley Data, we aim to uncover previously unrecognized off-target sites within oncogenic regions. The diverse nature of populations within these datasets makes this project particularly important for advancing diversity, equity, and inclusion in the gene editing field. Our comprehensive analysis will improve understanding of how genetic diversity impacts gene editing outcomes, ultimately guiding the design of safer CRISPR-based therapies.
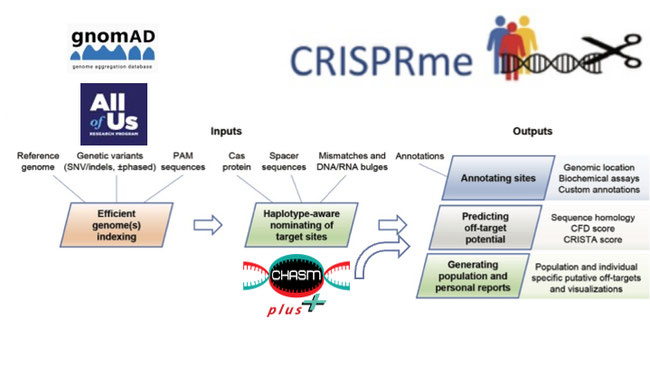
Our project aims to systematically identify potential off-targets (unintended editing events) of CRISPR gene editing that may contribute to oncogenesis. While many methods to nominate off-target events exist, they have key limitations. Our group recently developed CRISPRme[1], a variant and haplotype-aware tool that can enumerate potential off-targets based on reference populations to account for genetic diversity. CRISPRme introduces three key features lacking in other tools: (i) its aligner is specifically optimized for scanning guide RNAs (gRNAs), (ii) it accounts for DNA/RNA bulges, and (iii) it incorporates population genetic variation. CRISPRme has already demonstrated its effectiveness on smaller datasets (1000 genome project and Human Genetic Diversity Project or HGPD), yielding significant results, such as discovering an off-target site for a guide currently used in clinical trials and already approved by the FDA called Casgevy[7]. Casgevy is a sickle cell disease treatment, and the off-target we discovered is created by a variant observed in ~5% of individuals of african ancestry, the most affected population by sickle cell disease[1].
Given the demonstrated importance and impact of these analyses, we propose to extend CRISPRme capability by leveraging larger and more recent reference panels: the All of Us (AoU) Research Program[2], a US-driven initiative with the ambitious goal of gathering health data and genetic information from diverse populations, and the Genome Aggregation Database (gnomAD), a large collection of genetic variants from nearly a million individuals[3]. To assess risk of the enumerated off-targets we will also integrate these datasets with genes containing known cancer driver mutations from the CHASMplus dataset (hosted on Mendeley Data), to assess the risk of modifying these sites in relation to cancer development[4]. The rationale is that if a gene contains driver mutations, off-target editing in that gene could potentially contribute to oncogenesis. This intersection will also enable us to produce a simple yet powerful initial output: a detailed breakdown of variants in oncogenic genes, categorized by ethnicity and variant type.
Our timeline involves several key steps:
Month 1: Identify variants that intersect potentially oncogenic genes from CHASMplus.
Months 1–3: Use CRISPRme to predict potential off-target effects for multiple guide RNAs, currently used in clinical settings and therapeutic settings. Work to optimize CRISPRme and integrate the AoU and gnomAD datasets.
Months 3–6: Interpret the results and draft a manuscript for publication. Polish the new update of CRISPRme and make it more user friendly.
By providing deeper insights into how genetic diversity influences CRISPR off-target effects and oncogenesis risk and designing a more advanced computational tool, we aim to inform the design of safer CRISPR therapeutics, ultimately improving patient outcomes and advancing personalized medicine.
We expect to identify novel potential off-target sites for CRISPR gene editing that overlap with known oncogenic genes, particularly those that have been overlooked due to genetic diversity. By integrating genetic variant data from AoU and gnomAD with cancer driver mutations from the CHASMplus dataset, our comprehensive analysis will enhance understanding of risks associated with CRISPR-based therapies in diverse populations and in specific individuals.
In addition to performing off-target oncogenesis analysis we plan to release an updated CRISPRme. The original CRISPRme paper has already been cited 64 times and is used in both academia and industry for off-target search, the addition of AoU and gnomAD into CRISPRme as well as the optimization of runtime will be a great benefit to the field of genetic editing.[1]
In alignment with the FAIR (Findable, Accessible, Interoperable, Reusable) principles, we will share our processed datasets, analysis scripts, and pipeline through open-access repositories like Mendeley Data or Figshare, both part of the GREI. We will assign digital object identifiers (DOIs) and provide rich metadata to ensure that our data and tools are easily discoverable and accessible. By using standardized file formats and ontologies, we enhance interoperability and will license our data under terms that allow for reuse and redistribution, fostering further research and collaboration.
Regarding the CARE (Collective Benefit, Authority to Control, Responsibility, Ethics) principles, we will engage with data governance bodies to ensure the ethical use of genomic data, particularly respecting the rights and privacy of individuals represented in the AoU dataset. Our focus on datasets rich in genetic diversity, such as AoU and gnomAD, promotes diversity, equity, and inclusion (DEI) in genetic therapy. By ensuring that the benefits and risks of CRISPR-based treatments are assessed across different genetic backgrounds, we address health disparities and promote equitable healthcare outcomes.
To ensure replicability and reproducibility, we will provide comprehensive documentation of our methodologies and publish the updated version of CRISPRme on GitHub and also in a more user-friendly website version.
Importantly, this project has immediate implications for ongoing clinical trials based on CRISPR gene editing in various diseases, ranging from sickle cell and β-thalassemia to retinal disorders and cancer. It aligns with the FDA's recent draft guidance on gene therapy products[8], suggesting that considerations of human genetic diversity could be incorporated into final guidelines. Such alignment could influence everything from clinical trial design to market authorization and insurance reimbursement, potentially limiting access but enhancing safety.
We believe that our systematic off-target characterization leveraging available yet unutilized datasets has profound implications for the fields of gene editing, oncology, and personalized medicine. Recognizing how genetic diversity influences gene editing outcomes paves the way for more customized therapies, potentially leading to reduced side effects and heightened treatment efficacy for various genetic disorders. Moreover, this research enriches the broader understanding of the potentials and constraints of gene editing. By acknowledging the role of genetic diversity, the medical community can work towards more inclusive and effective therapies that benefit a broader range of people.
Discovering new potential off-target oncogenic sites has serious implications for the safety of CRISPR-based therapies. Identifying these sites allows us to design guide RNAs that minimize the risk of inducing harmful mutations, particularly those that could activate oncogenes or disrupt tumor suppressor genes. This is crucial for developing safe gene therapies for various diseases, ensuring that treatments do not inadvertently increase cancer risk.
There is an urgency to our analyses as therapies which are currently being used or developed might have potential off-target effects previously unseen and it is of the utmost importance for both scientific progress and public health that these potential oncogenic off-targets are discovered, quantified, and published for public use.
Our previous research discovered an off-target effect of a BCL11A enhancer targeting guide RNA which disproportionately affected people in African-ancestry populations. With incredibly more rich and diverse datasets this project promises to build on our previous work and continue to promote diversity equity and inclusion in treatment with CRISPR therapies.[1]
The shift from using a dataset such as 1000 genomes which has 88 million variants to the population data which is contained in AoU and gnomAD (which have over 1.5 billion variants combined) is not just a quantitative improvement but a qualitative one[2][3]. By enhancing CRISPRme to include data from AoU and gnomAD, we provide the scientific community with an improved tool capable of comprehensive off-target predictions that account for global genetic diversity.
The scientific significance of this project lies in its dual focus on discovering potential new off-target oncogenic sites and improving CRISPRme to handle larger and more diverse population genomic datasets. By advancing our understanding of how genetic diversity influences gene-editing outcomes and providing practical tools for the scientific community, we aim to pave the way for safer and more effective gene therapies. Our work promotes diversity, equity, and inclusion in genetic therapy by ensuring that the benefits and risks of CRISPR-based treatments are evaluated across different genetic backgrounds, ultimately improving patient care and advancing human health.
Our team consists of four members Manuel Tognon, Taylor Hudson, Ben Vyshedskiy, and Luca Pinello. We all work in the Pinello lab associated with Mass General Hospital(MGH), Harvard Medical School(HMS), and the Broad Institute.
Manuel Tognon is a Post-doc researcher at the University of Verona. Manuel's research focuses on developing cutting-edge computational tools to explore and predict the impact of genetic variants on epigenetic elements and CRISPR genome editing experiments outcome, with a focus on off-target nomination. Manuel was instrumental for the development of CRISPRme.
Taylor Hudson is a Research Technician at MGH and a current Ph.D. student in the Department of Molecular Therapeutics at the University of California, Berkeley. Her primary research focuses on leveraging tools for off-target prediction and screening. She has utilized CRISPRme to support clinicians at UCSF, UCB, and UPenn in advancing IND-enabling gene therapy trials.
Ben Vyshedskiy is a student at the University of Cambridge and an intern at the Pinello Lab. He has a strong background in bioinformatics and statistics with previous computational biology internships at HMS and Verve therapeutics.
Luca Pinello is an Associate Professor at Massachusetts General Hospital and Harvard Medical School and an Associate Member of the Broad Institute. His lab is focused on developing computational and machine learning tools to understand gene regulation and in support of genome editing and genomics assays.
Receiving this prize will be transformative for this project since it will enable us to secure important resources and give visibility to an underappreciated problem. To ensure the success of our project if given the prize we have addressed several key considerations. First, we have secured access to the necessary genomic datasets from the AoU Research Program and gnomAD, along with the cancer driver mutation data from Mendeley Data. Second, recognizing the computational intensity of running CRISPRme analyses on extensive datasets we have arranged for adequate computational infrastructure. Our team possesses the requisite expertise in bioinformatics, genomics, computing infrastructure and the utilization of CRISPRme, enabling efficient data processing and analysis. We have and will continue to strictly adhere to all guidelines to protect participant confidentiality. These considerations ensure that the project is well-prepared to achieve its objectives.
